













WhatsApp (принадлежит корпорации Meta, признанной экстремисткой и запрещённой в России) для Windows превратился в прожорливый тормоз, пользователи жалуются на гигабайты памяти и лаги. Пока Meta рассказывает об успехах WhatsApp с его 3 миллиардами пользователей, владельцы Windows-компьютеров всё чаще задаются другим вопросом: как мессенджер умудрился стать настолько медленным?
Журналист Windows Latest опубликовал разгромный материал о текущей версии WhatsApp для Windows, которую Meta перевела на WebView2 — фактически браузерную оболочку на базе Chromium.
По его словам, приложение способно потреблять сотни мегабайт оперативной памяти ещё до аутентификации пользователя. А после входа в аккаунт и активной работы объём занятой памяти легко переваливает за 1 ГБ.
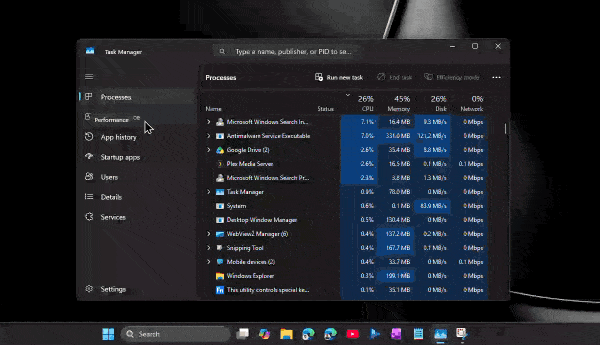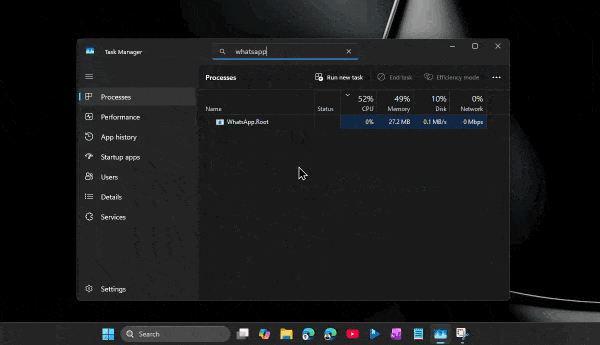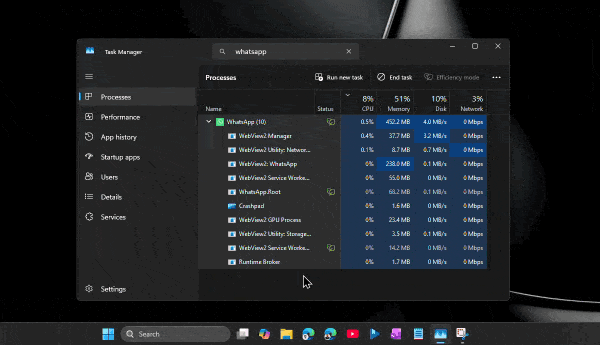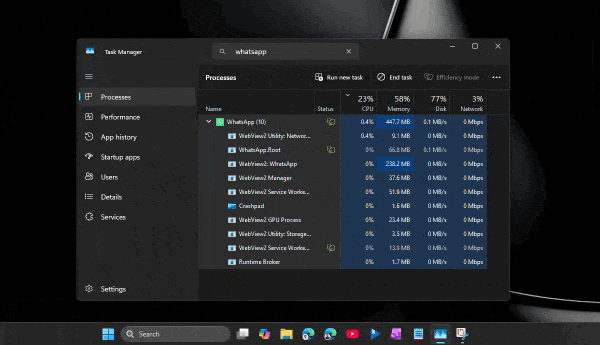
При этом проблема не только в аппетитах. Пользователи жалуются на задержки при отправке сообщений, медленное переключение между чатами, подтормаживания интерфейса и нестабильную работу после выхода компьютера из режима сна. Автор утверждает, что браузерная версия WhatsApp в обычной вкладке зачастую работает быстрее, чем официальное настольное приложение.
Особенно тяжело приходится владельцам старых ПК. В качестве примера приводится компьютер с процессором Intel Core i3 шестого поколения и 8 ГБ оперативной памяти. По наблюдениям автора, Windows 11 на такой машине работает вполне бодро, а вот WhatsApp способен загружать процессор более чем на 20 % даже в состоянии простоя.
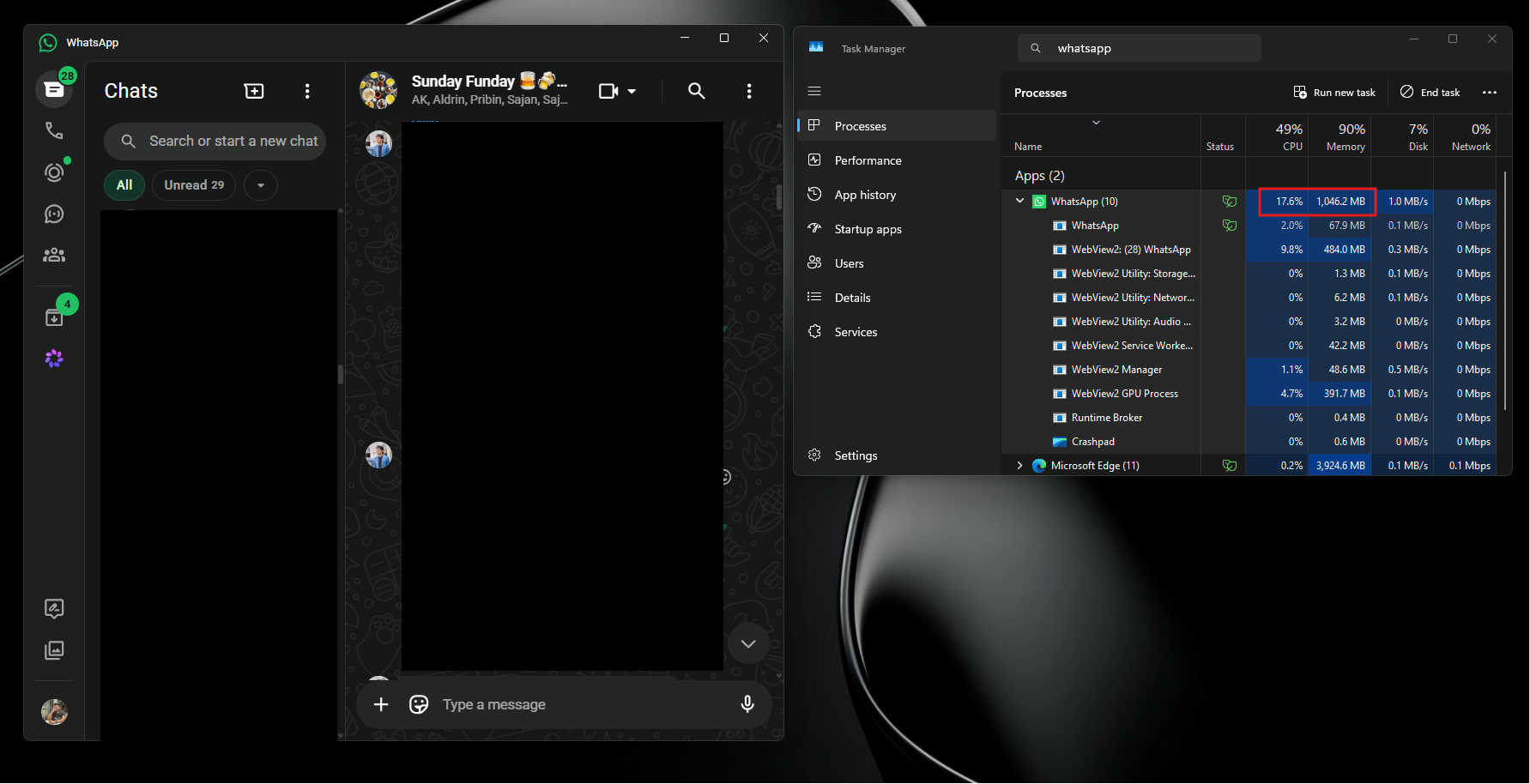
Главным виновником называется переход с нативного UWP-приложения на WebView2. Старая версия WhatsApp для Windows занимала менее 100 МБ памяти и использовала системные механизмы уведомлений. Новая же фактически запускает внутри себя мини-браузер с набором отдельных процессов для рендеринга, сети, хранения данных и других задач.
Досталось и Microsoft. Автор напоминает, что даже корпоративный Teams до сих пор построен на WebView2 и также известен высоким потреблением ресурсов. По его мнению, именно многолетняя непоследовательность Microsoft в развитии собственных платформ заставила разработчиков всё чаще выбирать веб-оболочки вместо нативных приложений.
Самое ироничное в этой истории — у Meta есть полноценный и хорошо оптимизированный WhatsApp для macOS и даже для Apple Watch. А вот для полутора миллиардов пользователей Windows компания предлагает по сути браузерную вкладку, упакованную в отдельное окно и выдаваемую за полноценное приложение.
Напомним, ранее мы рассказывали о способе вернуть быстрый нативный WhatsApp в Windows 11. Речь идёт о версии 2.2546.3.0 от ноября 2025 года — это ещё тот самый UWP-клиент. Если установить её вручную и немного подправить, Windows 11 позволит продолжить пользоваться нативным приложением.



